The globally pandemic coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). As of mid-October 2021, that catastrophic virus has infected an estimated 220 million people, and claimed more than 4.5 million victims. Since the beginning of COVID-19 outbreak, there is an urgent demand to identify the etiological factors and the molecular biological basis that contribute to its rapid spread, and severe morbidity and mortality of contracted patients. Recently, insightful pieces of data collected from sequence analyses of the viral genome and structure determination of the viral proteins by cryo-EM and X-ray crystallography dissect the molecular dynamics of SARS-CoV-2 pathogenesis. These virological discoveries greatly facilitate strategies to develop vaccines and treatments preventing disease transmission and exacerbation.
|
The spike (S) protein and how it mediates cell entry of SARS-CoV-2 |
A critical initial step of virus infection is attaching to receptors on host cells. Cellular entry of coronaviruses can be roughly divided into two steps: first, the binding of S protein to a specific surface receptor; then S protein is cleaved by cellular proteases to promote membrane fusion and viral uptake. SARS-CoV-2 principally employs angiotensin-converting enzyme 2 (ACE2) as receptor and transmembrane serine protease 2 (TMPRSS2) with ubiquitous furin as cooperative proteases for entry into cells. Immunohistochemistry and RNA-sequencing studies observe that epithelium cells of airway and gastrointestinal tract express high levels of ACE2 and TMPRSS2.
Dynamic landscape mapping the interacting characteristics of the receptor binding event of SARS-CoV-2 (Figure 1) reveal that S proteins exist as trimers in a metastable state on newly packaged virion where they fluctuate between distinct conformations. S protein contains two functional subunits, termed “S1” and “S2” (Figure 2). The receptor-binding domain (RBD), locating in globular S1 head, exhibits an affinity in the low nanomolar range to ACE2. In prefusion state, the flexible S1 RBD can perform hinge-like motions that switches between a dominant “down/closed” conformation when its RBM (receptor-binding motif) is inaccessibly buried and a transient “up/open” conformation, corresponding to an exposed RBM region.
ACE2 binding triggers an overall state shifts of S trimers toward postfusion. Compared to S protein of the previous SARS-CoV, that of SARS-CoV-2 acquired a furin-like protease cleavage site at the S1/S2 interface. Upon receptor engagement, the S1 domain is dissociated from the virion surface due to furin proteolytic activity. Such priming cleavage subsequently leads to further process of stalk-shape S2 domain by TMPRSS2 at the S2’ site, then cleaved S2 quickly refolding into stable postfusion state. The S2, itself a machinery set, is constituted by six motifs required drive lipid bilayers fusion. The second cleavage initiates the fusion machinery and afterwards the hydrophobic fusion peptide (FP) embeds into the host cell membrane. During this dramatic structure rearrangement of a S2 trimer, a six-helix bundle (6HB) is formed by antiparallelly associating two heptad repeat (HR1 and HR2) motifs. Multiple 6HBs act in concert to conduct close juxtaposition of host cell membrane intercalated with FR and viral envelope anchored by transmembrane (TM) regions and later opening of a small lipid fusion pore which interconnecting both ends. Through the expanding pore, pathogenic virus components such as nucleocapsid and ORF1ab polyprotein reach intracellular space.
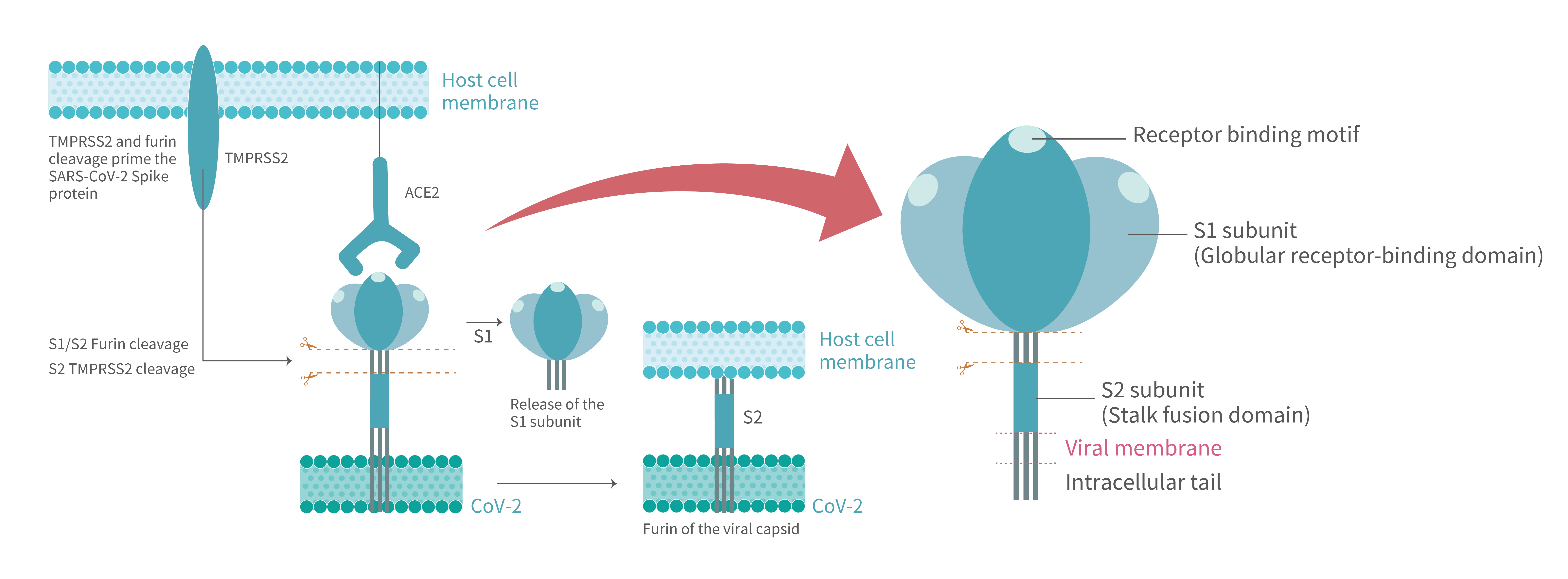 Figure 1. After SARS-CoV-2 S protein binds to the ACE2 receptor on host cell, where the viral entry process is primed by the cleavage of two protease,furin and TMPRSS2. Proteolytic activation of the spike trimer at the boundary sites between the S1 and S2 subunits dissociates the receptor-binding S1 domain and exposes the S2 domain required for mediating membrane fusion.
Figure 1. After SARS-CoV-2 S protein binds to the ACE2 receptor on host cell, where the viral entry process is primed by the cleavage of two protease,furin and TMPRSS2. Proteolytic activation of the spike trimer at the boundary sites between the S1 and S2 subunits dissociates the receptor-binding S1 domain and exposes the S2 domain required for mediating membrane fusion.
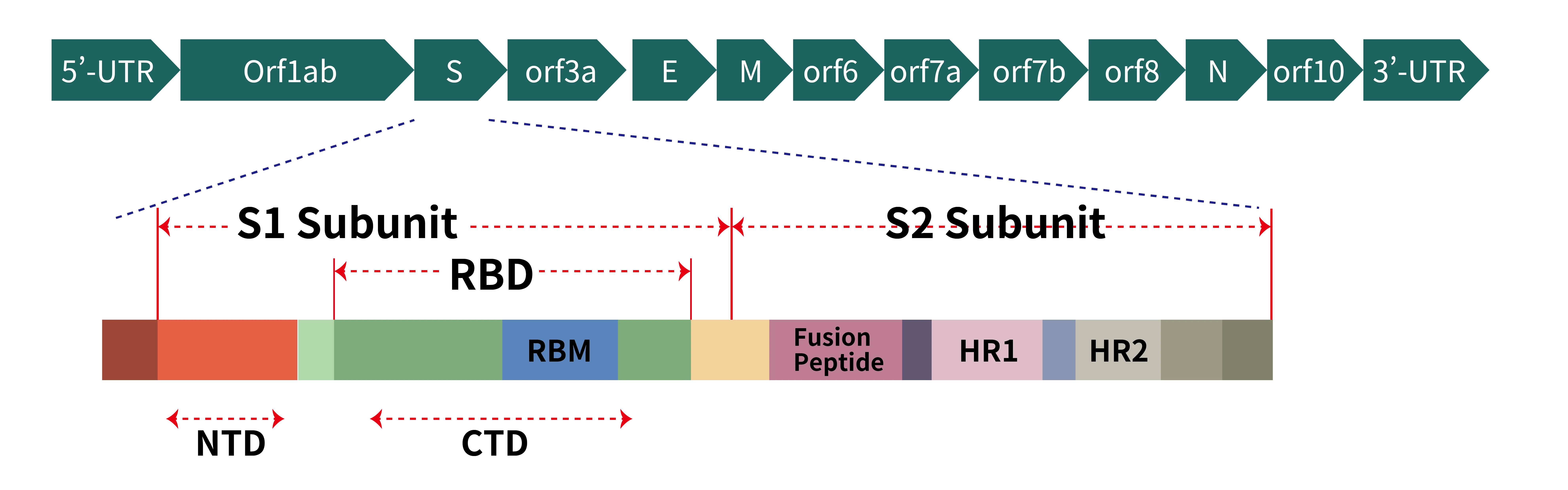 Figure 2. (a) Schematic representation of genome organization of SARS-CoV-2; (b) Schematic representation of the domain arrangement of the SARS-CoV-2 S proprotein. SS, signal peptide; NTD: N-terminal domain; RBD: receptor-binding domain; RBM: receptor-binding motif; SD1/2: subdomain 1 and 2; FP, fusion peptide; HR1, heptad repeat 1; CH, central helix; CD, connector domain; HR2, heptad repeat 2; TM, transmembrane domain; CT, cytoplasmic tail. Arrows denote protease cleavage sites. The SARS-CoV-2 S glycoprotein is synthesized as a 1273-amino acid polyprotein precursor.
Figure 2. (a) Schematic representation of genome organization of SARS-CoV-2; (b) Schematic representation of the domain arrangement of the SARS-CoV-2 S proprotein. SS, signal peptide; NTD: N-terminal domain; RBD: receptor-binding domain; RBM: receptor-binding motif; SD1/2: subdomain 1 and 2; FP, fusion peptide; HR1, heptad repeat 1; CH, central helix; CD, connector domain; HR2, heptad repeat 2; TM, transmembrane domain; CT, cytoplasmic tail. Arrows denote protease cleavage sites. The SARS-CoV-2 S glycoprotein is synthesized as a 1273-amino acid polyprotein precursor.
|
Immunopathological roles of the SARS-CoV-2 ORF8 (open reading frame 8) |
SARS-CoV-2 is a positive single-stranded RNA virus, whose genome of around 30 kb encodes for proteins that are common to all members of the coronavirus such as replicase polyproteins (pp1a and pp1b) and structural proteins (spike, membrane, nucleocapsid and envelope), to which many antiviral agents and prophylactic vaccines are being developed to target. However, there are also eleven accessory proteins (Figure 3), which have been designated as open reading frames (ORF) 3a, 3b, 3c, 3d, 6, 7a, 7b, 8, 9b, 9c and 10, and they share low sequence homology to viral proteins of other known coronaviruses. As obligate intracellular parasites, viruses may utilize several mechanisms to operate or evade the host immune system to establish a productive infection and maintain an optimal replicative niche. Although most accessory proteins are supposed not to directly involve in viral replication and invasion, accumulating evidence suggests that accessory proteins may act as virulence factors to suppress immune responses or intercept substance metabolism of host cells. Specifically, some natures of key players in host immunity are impaired by these accessory proteins.
Type I interferons (IFNs-I, often refers to IFN-α and IFN-β) are produced by almost every cell of the body in defense of virus infections. IFNs-I induce antiviral activities by inhibiting the proliferation of virus-infected cells and enhancing antigen presentation via major histocompatibility complex class I molecules (MHC-I) upregulation. In addition, IFNs-I mediate diverse interplays on innate and adaptive immune cells, and lead to expression of a gene collection known as IFN-stimulated genes (ISGs). Cytotoxic T lymphocytes (CTLs also called CD8+ T cells) can detect and destroy virus-infected cells by monitoring the presence of viral peptide antigens that are loaded to MHC-I and displayed on cell surface. Namely, cytolytic functionalities of CTLs are antigen-specific and restrict to MHC-I presentation. Some preliminary mechanisms have been proposed to explain how viral accessory proteins function as measure to counteract intracellular antigen presentation through hindering IFNs-I signaling and MHC-I assembling and trafficking.
SARS-CoV-2 ORF8 has 366 nucleotides and 121 amino acids. In the first 17 amino acids of its N-terminus, a signal peptide for endoplasmic reticulum (ER) import has been identified, but ORF8 is proved as a secreted protein and highly immunogenic in COVID-19 patients. ORF8 also possesses a domain with putatively immunoglobulin (Ig)-like fold which presumably interact with a variety of host proteins. In comparison to genes of other accessory proteins, ORF8 gene seems to be more susceptible to deletions and nucleotide substitutions, hence generating many variants that would potentially diversify interplay between viral proteins and host factors.
Recent studies showed that ORF8 may bind to MHC-Ι molecules and mediate their down-regulation. In ORF8-expressing cells, MHC-Ι molecules are selectively targeted and enriched in lysosomes for degradation. In severe COVID-19 patients, subverted immunity characterized with dysfunctional IFNs-I responses and persistently high viral loads. ORF8 might exert an IFN antagonism in cooperation with ORF6 and N protein. Taken together, immunomodulatory properties of ORF8 allows cells contracting with SARS-CoV-2 more resistant to recognition and attacks of immune cells.
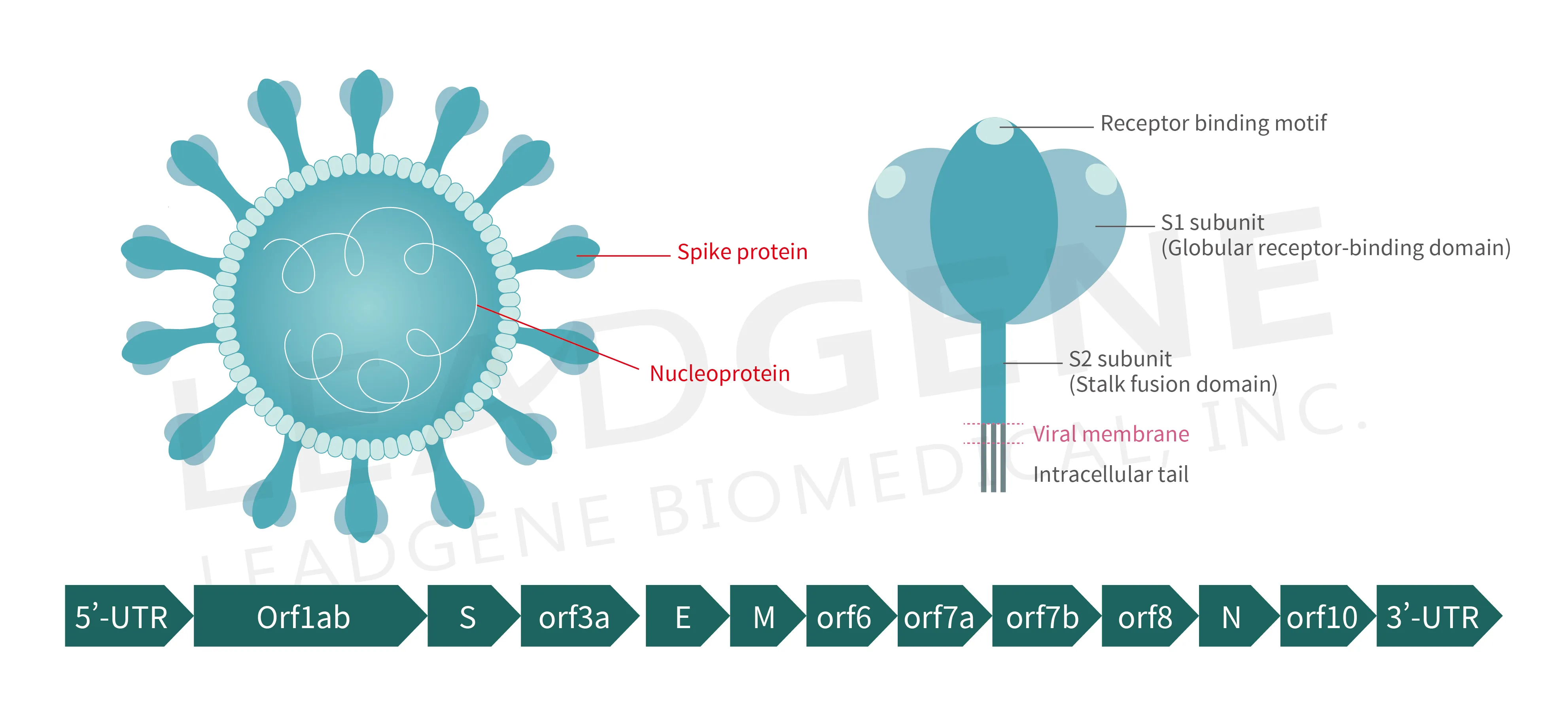 Figure 3. Schematic representation of the virion anatomy and genome organization of SARS-CoV-2
Figure 3. Schematic representation of the virion anatomy and genome organization of SARS-CoV-2
